Фенилкетонурия
Содержание:
- Диагностика
- Прогноз и профилактика
- Грудное вскармливание в ФКУ
- Применение препарата КУВАН при некоторых формах фенилкетонурии
- «Золотые» продукты
- Диагностика
- Лечение
- Создание казенного учреждения
- Диагностика фенилкетонурии
- Ответы на часто задаваемые вопросы
- Функции телефона и электронной почты организации
- Патогенез
- Лечение фенилкетонурии
- Как лечить фенилкетонурию
- История
Диагностика

Анализ крови на фенилкетонурию проводят каждому ребенку еще в роддоме. Чем раньше ребенку поставлен диагноз «фенилкетонурия», тем лучше прогноз и больше вероятность нормального развития интеллекта.
В связи с высокой распространенностью данной патологии, тяжестью течения и реальной возможностью профилактического лечения ВОЗ включила ее в список наследственных заболеваний, для выявления которых проводится массовый скрининг новорожденных. Все дети на 4-5-й день жизни должны обследоваться на фенилкетонурию. Для этого у ребенка производят забор крови, пропитывают ее каплей специальный бланк и отправляют на исследование в лабораторию медико-генетического центра. По результатам этого анализа (уровень фенилаланина) оценивают вероятность развития болезни у ребенка.
- При обнаружении стойкой гиперфенилаланинемии проводится дифференцирование классической формы болезни от атипичных.
- При незначительном увеличении концентрации этой аминокислоты в крови проводятся повторные анализы.
В более старшем возрасте предположить наличие заболевания у ребенка врач может на основании клинических данных. Особенно высокий риск его развития имеют дети, в семьях которых уже регистрировались случаи болезни. Подтвердить диагноз позволяют лабораторные исследования:
- определение уровня фенилаланина в крови (более 900 мкмоль/л);
- выявление его производных в моче (проба Феллинга);
- обнаружение дефектных генов с помощью молекулярно-генетических методов (полимеразно-цепная реакция).
Прогноз и профилактика
Предупредить развитие необратимых повреждений органов ЦНС позволяет ранняя диетотерапия. Для того чтобы лечебное питание было назначено вовремя детям практически сразу после рождения проводится массовый скрининг и при необходимости назначается дополнительная диагностика.
Своевременное проведение элиминационной диеты и неукоснительное соблюдение предписаний врача по лечебному питанию позволяет родителям вырастить полностью здорового ребенка, то есть без отклонений в интеллектуальном, физическом и психическом развитии.
Прогноз течения патологии неблагоприятный при поздно начатой диетотерапии. Если в подростковом возрасте диета расширяется неадекватно или ее соблюдение полностью прекращается, то наблюдается снижение способности к обучаемости, нарушение поведенческих норм, расстройства со стороны психики.
Вот почему большинство психотерапевтов рекомендуют придерживаться элиминационной диеты как минимум до 18 лет.
Риск рождения детей, больных фенилкетонурией, оценивается после обследования супружеских пар в медико-генетических центрах. Обязательно такому обследованию должны подвергаться супруги, у которых уже рожден ребенок с таким заболеванием.
Желательно чтобы при планировании зачатия обследование на генетические патологии проходили пары, имеющие близкое кровное родство.
При атипичных формах ФКУ диетотерапия нужных результатов коррекции расщепления фенилаланина не дает. Прогноз течения таких типов патологии неутешительный – малыши либо гибнут в первые годы рождения, либо у них наблюдается глубокая умственная отсталость.
Фенилкетонурия пока единственное наследственное заболевание, раннее лечение которого предупреждает практически на 100 процентов вероятность развития тяжелых осложнений в будущем.
Поэтому родителям нельзя отказываться от неонатального скрининга их малыша в роддоме, а при рождении ребенка дома нужно его обследовать в течение первых трех недель жизни.
Грудное вскармливание в ФКУ
Ребёнка с ФКУ можно кормить грудью, но с ограничением. Существуют два способа кормления грудных детей. I способ: можно кормить поочерёдно, что требует взвешивания ребёнка до и после кормления: a) одно кормление только грудным молоком, а b) следующее кормление бутылка с приготовленным препаратом ФКУ. II способ: каждое кормление можно поделить на две части: a) сначала кормим ребёнка строго рассщитанным количеством препарата, b) a затем докармливаем грудным молоком до сытости. Количество поступившего в организм грудного молока рассчитываем взвешивая ребёнка до и после кормления. Если нет возможности контролировать количество грудного молока выбираем III способ: молоко должно быть сцежено и подано в объёме, не превышающем рекомендации вместе с соответствующим количеством лечебной смеси.
В случае отсутствия грудного молока источником фенилаланина в диете может быть использовано детское питание для грудных детей (напр. Bebilon 1, Bebiko 1). Суточная потребность ребёнка в молочной смеси зависит от его индивидуальной толерантности фенилаланина и должна быть в точности рассчитана. Остальное количество белка без фенилаланина вместе с другими питательными компонентами поступает из препарата ФКУ.
Голодный ребёнок Ребёнок, который проявляет симптомы голода, а уже употребил суточную норму фенилаланина с грудным молоком или молочной смесью, может получить дополнительную дозу питания без фенилаланина в форме препарата ФКУ стандартно разбавленного.
3 мерки препарата + 90 мл воды = 100 мл готовой смеси
Применение препарата КУВАН при некоторых формах фенилкетонурии
По материалам IV Международного медицинского форума, Киев, 16-18 апреля 2013 года. Лекции профессора Trefz Friedrich (Германия), Пичкур Н.А. (Метаболический центр ДКЛ «ОХМАТДЕТ», Киев)
Соблюдение диеты при ФКУ позволяет добиться эффективного лечения, но представляет сложность для пациентов, так как ограничивает их социальную активность, особенно в подростковом возрасте и у взрослых, что приводит к частому отказу от диеты.
Существуют некоторые формы ФКУ, при которых вместо традиционной диетотерапии возможно применение препарата КУВАН (сапроптерина гидрохлорида), который усиливает остаточную активность фенилаланингидроксилазы (РАН*), таким образом снижая уровень ФА. Другими словами, КУВАН стимулирует активность фермента, под действием которого фенилаланин превращается в тирозин.
Из всех наблюдаемых пациентов с ФКУ
- 50% — это классическая ФКУ,
- 20% — это нефенилкетонурическая ГФА (не РАН-ГФА*)
- 30% — мягкая ФКУ
Однако, не все пациенты с ФКУ чувствительны к терапии КУВАНом. Как правило, пациенты с классической ФКУ не чувствительны, а примерно 35 % от общего числа пациентов с нефенилкетонурической ГФА и мягкой ФКУ могут применять КУВАН.
«Золотые» продукты
Согласно постановлению правительства №890, пациенты с ФКУ имеют право на бесплатное предоставление как аминокислотной смеси, так и низкобелковых продуктов.
«Но в реальности выдают только смеси, а продукты — на усмотрение региона. С 2020 года в Кемеровской области выдают низкобелковые продукты, за что мы очень благодарны главному внештатному генетику и администрации. Но таких регионов по РФ немного. Остальным родителям детей с ФКУ и взрослым пациентам приходится покупать продукты самостоятельно», — рассказывает Елена Мареченкова.
Такие цены обусловлены в том числе и тем, что большинство продуктов, которые едят взрослые и дети с ФКУ, — импортные. «Заказываем из Германии, Польши, — рассказывает Матлюба Хакимова. — Потому что их продукты вкуснее, почти не отличаются от обычных по консистенции, вкусу».
Есть и российские производители, они дешевле. «Если наши макароны я могу купить за 100 рублей 400 г, то зарубежные будут уже 400—500 рублей за те же 400 г. Но нужно сказать, что наш продукт уступает по качеству импортному», — объясняет Хакимова.
Еду для людей с фенилкетонурией готовить непросто, добавляет Елена Мареченкова: «Нужно тщательно взвешивать все продукты, записывать, сколько ребёнок съел за день. К тому же низкобелковую пищу надо ещё уметь приготовить».
«Не одна упаковка продуктов уходит в мусорку, прежде чем родитель поймёт, как с ней работать. У меня как-то ушла 200-граммовая пачка заменителя яйца за 1,5 тыс. рублей, потому что я никак не могла понять, как сделать из этого омлет, получался какой-то кисель застывший», — рассказывает Татьяна Слепцова, мама ФКУ-ребёнка и руководитель региональной общественной организации помощи людям с фенилкетонурией по Саратовской области «Вкус жизни».
Диагностика
Важным, как мы уже отметили, является раннее диагностирование заболевания, что позволит избежать его развития и привести к ряду необратимых и тяжелых последствий. По этой причине в родильных домах к 4-5 дням жизни (для новорожденных доношенных) берется для анализа кровь. У недоношенных детей на предмет фенилкетонурии (ФКУ) кровь берется на 7 день.
Процедура предусматривает взятие капиллярной крови по прошествии часа с момента кормления, ею в частности пропитывается специальный бланк. Концентрация, указывающая на отметку свыше 2,2% фенилаланина в крови малыша, требует направления его с родителями для осмотра в медико-генетический центр. Там же проводится дообследование и, собственно, уточнение диагноза.
Лечение
Основа лечения заключается в назначении специализированной диеты. Необходимо провести просветительскую работу с родителями ребенка, убрать из сознания психологическую установку «наследственное – значит неизлечимое и смертельное»
Патология поддается терапии, особенно важно соблюдение всех установок к лечению в первые дни жизни ребенка. Диета направлена на снижение, но не полное исключение фенилаланина
Всю жизнь желателен контроль уровня фенилаланина в плазме крови. Оптимальным считается уровень в 1-1,8 ммоль/л.
В пищу на первых днях жизни используются специальные смеси (афенилак, нутриген и т.д.), также допустимы овощные и фруктовые пюре. Молоко из пищи исключается. В дальнейшем показано употребление большинства овощей (огурцов, моркови, капусты, помидор,), фруктов (яблок, винограда, апельсинов) и жесткое ограничение продуктов животного происхождения. Допустимы мед, ягодные варенья.
С возрастом проницаемость гематоэнцефалического барьера для аминокислот снижается. В силу этого уменьшается и токсическое воздействие фенилаланина на мозг, и контроль уровня аминокислоты в крови можно проводить гораздо реже.
Важным вопросом является профилактика заболевания и планирование семьи. Гетерозиготных носителей дефектных генов (а их встречаемость в среднем среди населения достигает 1,5-2%) можно выявить при проведении специальных нагрузочных проб. Целесообразно проводить такое исследование при наличии в наследственном анамнезе случаев фенилкетонурии и родственников первых линий родства.
Создание казенного учреждения
Такой тип государственных учреждений создается по распоряжению федерального или регионального органа исполнительной власти, муниципалитета (ч. 2 ст. 13 закона № 7-ФЗ).
Обратите внимание! На федеральном уровне этот процесс регламентирован Порядком создания, реорганизации, изменения типа, ликвидации…, утв. постановлением Правительства РФ от 26.07.2010 № 539
На уровне субъекта РФ или муниципалитета действуют уже свои акты.
Пример — Порядок, утв. постановлением правительства Тюменской области от 28.12.2010 № 393-П, и Порядок, утв. постановлением администрации Заводоуковского городского округа от 30.11.2010 № 1887.
Обычно процесс создания казенного учреждения включает следующие этапы:
- Принятие учредителем решения о создании юрлица.
- Оформление проекта распоряжения/постановления о создании учреждения. Он, как правило, содержит:
- название учреждения, его тип;
- основные виды и цели деятельности, функции казенного учреждения, разрешенные для данного казенного учреждения платные услуги и т. д.;
- наименование учредителя;
- перечень мероприятий, направленных на создание учреждения, с обозначением их сроков и ответственных за их реализацию структур.
- Составление пояснительной записки к проекту с обоснованием целесообразности создания учреждения.
- Согласование проекта с ответственными подразделениями учредителя (например, департаментом имущественных отношений и правовым департаментом).
- Издание постановления о создании учреждения.
- Утверждение устава учреждения путем издания распоряжения/ постановления.
- Направление документов в ФНС для государственной регистрации юрлица.
Рекомендуем! Как направить документы для госрегистрации юрлица через портал «Госуслуги», читайте в нашей статье «Регистрация юридического лица на портале госуслуг».
Устав
Учредительный документ казенного учреждения — устав — содержит сведения:
- о названии юрлица, его типе (в названии учреждения обязательно должно быть указано, что оно казенное);
- местонахождении юрлица;
- учредителе и собственнике имущества;
- видах деятельности, организации, управлении юрлицом, его структуре и т. п.;
- имуществе и финансовом состоянии, в т. ч. порядке распоряжения имуществом, его передаче другим учреждениям, совершении крупных сделок и сделок с заинтересованностью и т. д.;
- обособленных подразделениях юрлица и т. д.
Обратите внимание! Устав утверждает учредитель юрлица посредством принятия правового акта. Порядок согласования, утверждения и внесения изменений в устав обычно регламентирован
Например, для устава федерального государственного казенного учреждения порядок утвержден постановлением Правительства РФ от 26.07.2010 № 539.
Руководитель
Единоличный исполнительный орган управления казенного учреждения — директор/руководитель. Его назначает собственник имущества учреждения, издав специальное распоряжение. Директора выбирают посредством проведения конкурса. Он подотчетен собственнику имущества.
Директор выполняет функции по организации деятельности учреждения в соответствии с целями последнего, совершает сделки от его имени, решает кадровые, административно-хозяйственные и прочие вопросы, возникающие в процессе работы юрлица.
Имущество казенного учреждения
Рекомендуем! О том, как зарегистрировать право оперативного управления на имущество, читайте в нашей статье «Государственная регистрация права оперативного управления на объекты недвижимости».
Диагностика фенилкетонурии
 Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия — самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии. К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Министерства охраны здоровья организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории Украины.
Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия — самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии. К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Министерства охраны здоровья организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории Украины.
Скрининг-тест производится не раньше чем на 3 сутки (72 часа) после рождения ребенка. При обнаружении у ребёнка повышенного уровня фенилаланина (гиперфенилаланемии) скриниговая лаборатория согласовывает тактику в отношении каждого пациента с ведущим врачом-генетиком.
При уровне ФА в скриниге:
- 3-8мг% — родителей или опекунов информируют о необходимости выполнения контрольного исследования (колориметрическим методом). Если в котрольном исследовании уровень ФА выше 2мг%, то необходимо оповестить родителей/опекунов и пригласить в письменной форме в специализированный диагностический лечебный центр;
- выше 8мг% — необходимо оповестить родителей/опекунов о результате скринигового теста и пригласить в письменной форме в специализированный диагностический лечебный центр.
Во время первого визита врач-специалист (чаще всего педиатр или генетик) обязан предоставить родителям/опекунам исчерпывающую информацию относительно предполагаемого заболевания, его причин и возможностей лечения.
В связи с существованием ряда причин появления гиперфенилаланемии (фенилкетонурия, нетипичные формы фенилкетонурии и тирозенемия), необходимо провести дифференциальную диагностику. Если подтвердится дефицит гидроксилазы фенилаланина (фенилкетонурия), применяется диетотерапия. В случае, если причиной окажется нарушение обмена веществ иного рода, будут избраны иные методы лечения.
Типы фенилкетонурии
Классическая фенилкетонурия –phe > 1200 μмол/л (20 мг%) Умеренная фенилкетонурия – phe – 900 -1200 μмол/л (15 — 20 мг%) Мягкая фенилкетонурия – phe –600–900 μмол/л (10-15мг%) Мягкая гиперфенилаланинемия (gray zone) — phe – 360 – 600 μмол/л (6 — 10мг%) Мягкая гиперфенилаланинемия- не нуждающаяся в лечении – phe – 120 – 360 μмол/л (2 — 6мг%) (NIH PKU Conference report: State of the science and future research needs. Feb.22-23.2012) Злокачественная гиперфенилаланинемия – недостаточность тетpагидpобиоптеpина (BH4)
Дефицит гидроксилазы фенилаланина (PAH)
— фенилаланин ≤ 7 мг% (6 мг%) не нуждается в лечении – наблюдение!- фенилаланин > 7 мг% (6 мг%) – низкофенилаланиновая диета
Дефицит BH4 (злокачесивенная ФКУ) — фармакологическое лечение.
Клинические появления злокачественной фенилкетонурии:
— тяжелые и быстро прогрессирующие неврологические нарушения (несмотрия на нормальный уровень ФА в крови вследстве диетотерапии)- гипотония- спастический синдром- атаксия- нарушение глотания- беспокойство- прогрессирующая умственная отсталость- тяжело купирующиеся приступы судорог – наиболее характерные (до 3 мес. жизни признаки заболевания могут отсутствовать)- уровни фенилаланина нехарактерные ( высокие – как в классической ФКУ или низкие — как в мягкой гиперфенилаланинемии).
Прогрессирующее повреждение нервной системы при злокачественной фенилкетонурии может вести к смерти ребенка, если заболевание не будет обнаружено и не будет применено соответствующее лечение.
Также о дифференцированной диагностике читайте в статье Рекомендации по лечению фенилкетонурии в Польше
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Функции телефона и электронной почты организации
Вопросы, на которые предоставляют ответы в организации, могут быть исключительно информационными – данное учреждение не занимается инспекторскими функциями. Это отвечает сути открытия такого сервиса – чтобы налоговая служба не занималась лишней работой – информационным сопровождением и наполнением электронных баз разной документацией. На дворе давно 21-й век, имеются все современные электронные технологии – другими словами, произошли реформы информационного характера, через которые на данный момент и проходит российская система налогообложения.
Качество деятельности изучаемого нами сервиса — это важнейший показатель любого филиала налоговой службы. Если есть много положительных отзывов – налоговое администрирование проводится на высоком уровне. В ином случае имеются серьезные пробелы, которые отлично выявляются при анализе деятельности данного учреждения. Допустим, московская организация помогла серьезно упростить взаимодействие налогоплательщика с ФНС Москвы. Это привело к обеспечению своевременной отсылки сообщений, к повышению числа собранных сборов.
Надеемся, что изложенная информация помогла вам разобраться в том, что представляет собой сервис ФКУ – «Налог – Сервис» и разъяснила более детально принципы и суть его работы. Подводя итоги, можно отметить следующее: организация хорошо помогает простым гражданам разобраться с вопросами информационного и технического характера, не напрягая ими специалистов налоговых органов.
Патогенез
При фенилкетонурии в печени больного не продуцируется особый фермент под названием фенилаланин-4-гидроксилаза.
Основная его функция – преобразование поступающего в органы ЖКТ с пищей фенилаланина в аминокислоту тирозин. Это аминокислота входит в состав большинства ферментов, гормонов, способствует выработке меланина (пигмент), принимает участие в образовании белков и в функционировании большинства внутренних органов.
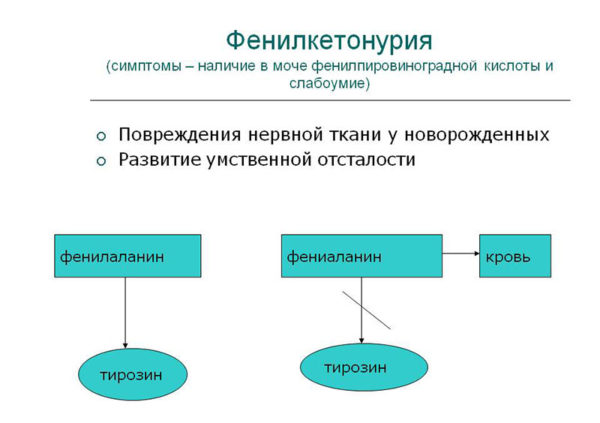
Метаболический блок при фенилкетонурии неуклонно приводит к тому, что начинают работать побочные (атипичные) пути обмена фенилаланина, в результате он трансформируется в те вещества, которых в теле здорового человека быть не должно.
Это кислоты – фенилмолочная и фенилпировиноградная, ортофенилацетат, фенилэтиламин. По сути, они являются токсическими соединениями, и их накапливание в кровеносной системе приводят:
- К нарушению нормального обмена липидов (жировых клеток) в разных отделах головного мозга;
- К нехватке нейромедиаторов, ответственных за бесперебойную передачу нервных импульсов во всей нервной системе.
В результате это приводит к прогрессирующему снижению интеллекта и к умственной отсталости выраженной степени – олигофрении, имбецильности, идиотии.
Отсутствие терапии становится причиной того, что через каждые 10 недель коэффициент развития интеллекта больного малыша падает на 5 пунктов.

Эхолалия – методы лечения, возможные последствия заболевания
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека
Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Как лечить фенилкетонурию
Единственным действенным методом лечения фенилкетонурии считается организованная с первых дней жизни специально разработанная диета, принцип которой заключается в ограничении содержащегося в продуктах питания фенилаланина, для чего исключаются такие продукты питания как:
- крупы,
- бобовые,
- яйца,
- творог,
- хлебобулочные изделия,
- орехи,
- шоколад,
- рыба, мясо и пр.
Лечебный рацион больных фенилкетонурией состоит из специализированных продуктов как зарубежного, так и отечественного производства. Детям первого года жизни показаны продукты, по своему составу приближенные к грудному молоку, это такие смеси как «Лофенилак» и «Афенилак». Для детей немного старше разработаны такие смеси как «Тетрафен», «Максамум-ХР», «Фенил-Фри». Страдающим фенилкетонурией беременным женщинам и детям старшего возраста (после шести лет) показан прием смеси «Максамум-ХР». Помимо специализированных лечебных продуктов в рацион больного включают соки, фрукты и овощи.
Находящиеся на лечении дети должны находиться под неусыпным контролем психоневролога и участкового педиатра. В начале лечения фенилкетонурии контроль содержания фенилаланина проводят еженедельно, при нормализации показателей переходят на 1 раз в месяц в течение первого года жизни, и 1 раз в два месяца у детей старше года.
Кроме диетотерапии, детям с фенилкетонурией врачи могут делать следующие назначения:
- минеральные соединения;
- ноотропные средства;
- витамины группы В;
- антиконвульсанты.
В комплексной терапии должны присутствовать лечебная физкультура, иглорефлексотерапия и массаж.
Обратите внимание: при атипичной форме фенилкетонурии, которая не поддается коррекции диетотерапией, врачи назначают гепатопротекторы, противосудорожные средства. Такое лечение поможет облегчить состояние ребенка
История
Фенилкетонурия как болезнь, протекающая с задержкой интеллектуального и психического развития, открыта в 1934 году норвежским ученым-исследователем Иваром Асбьером Феллингом, отсюда и другое обозначение патологии – болезнь Феллинга.
Первых успехов в коррекции здоровья малышей с ФКУ добились медики во главе с Хорстом Биккелем в середине прошлого века. Разработка терапии и ее внедрение проводились на базе Бирмингемского детского госпиталя (Англия).
Однако больших положительных результатов ученым удалось добиться, когда начала широко проводится ранняя диагностика новорожденных на выявление фенилкетонурии, это примерно 1958-1961 год прошлого века.
Расширение возможностей диагностики со временем позволило установить, что в развитии заболевания принимает участие только ген фенилаланингидроксилазы (РАН).
За последние десятилетия описаны атипичные варианты течения болезни, разработаны и широко применяются новейшие способы ее коррекции. В перспективе – использование генотерапии, что вполне вероятно позволит полностью победить болезнь.